Myelodysplastic syndromes (MDS)
MDS can be grouped into two major categories: higher risk or lower risk.6 Patients with higher-risk MDS carry a major risk of progression to acute myeloid leukaemia and short survival.7 Lower-risk MDS account for the majority of patients with MDS with anaemia being the main clinical challenge.6,8
Red blood cell transfusions provide temporary management of severe anaemia, as patients who receive transfusions may continue to experience fluctuations in haemoglobin.9 Patients receiving red blood cell transfusions may also experience complications, such as iron overload and alloimmunisation.10,11
Treatment of β‑thalassaemia

Professor Maria Domenica Cappellini, Professor of Internal Medicine,
University of Milan
In this video, Professor Cappellini discusses the classification and treatment of transfusion-dependent and non-transfusion-dependent β‑thalassaemia.
6-minute video
Differences and similarities of MDS and β‑thalassaemia

Doctor Esther Natalie Oliva, Haematologist, Grande Ospedale Metropolitano
Bianchi-Melacrino-Morelli
In this video, Doctor Oliva introduces MDS and β‑thalassaemia and the differences between the diseases. Doctor Oliva also discusses the differences and similarities in the treatment of both diseases
8-minute video
Other haematological diseases
Aplastic anaemia
The goals of treatment for aplastic anaemia are to restore haematopoietic stem cells and ameliorate cytopenia-related complications.18 Treatment options may include stem cell transplant (the only treatment to offer a potential cure), immunosuppressive therapy, and stem cell stimulation therapy may be an option for patients refractory to or relapsing after immunosuppressive therapy and androgens.18-20 Blood transfusions (of platelets or red blood cells) and other supportive care measures are often utilised.18
Myelofibrosis
The current treatment goals for myelofibrosis are to reduce the burden of symptoms and reduce the risk of leukaemic transformation.21,22 For high-risk patients, treatment options may include stem cell transplant (the only treatment capable of extending survival or offering a potential cure) or an investigational agent within a clinical trial.21,22 For low-risk patients, observation alone may be an option and for intermediate-risk patients, and low-risk patients requiring treatment, options include investigational agents in the setting of a clinical trial.21 For patients not eligible for stem cell transplant or clinical trial enrolment, symptom-directed therapeutic options include conventional treatments for anaemia, splenomegaly, constitutional symptoms, localised bone pain, or symptomatic extramedullary haematopoiesis.21,22
Sickle cell anaemia
Treatment for sickle cell anaemia usually aims to relieve symptoms and prevent complications.23 Treatment may include red blood cell transfusions, antibiotics to treat infections, antibiotics as prophylaxis to prevent infections in infants, pain relieving medications and hydroxyurea to reduce the frequency of painful crises.23,24 Stem cell transplant is an option to provide a potential cure but is usually only offered to younger patients.23
Congenital dyserythropoietic anaemias
Congenital dyserythropoietic anaemias are generally managed using red blood cell transfusions and iron chelation therapy. As with β‑thalassaemia, the only definitive cure for patients with congenital dyserythropoietic anaemias is stem cell transplant, however, this is limited to patients with very severe anaemias.3,25
Inherited
sideroblastic
anaemias
For mild inherited sideroblastic anaemias a watch and wait approach may be adopted whilst patients with mild-to-severe anaemia may be treated with ESAs or regular red blood cell transfusions.2 Hypomethylating agents and stem cell transplant may be considered for the most severe cases.2
The limitations of red blood cell transfusions for chronic anaemia
Red blood cell transfusions for chronic anaemia in patients with haematological diseases can improve the symptoms of anaemia. However, red blood cell transfusions can also impact quality of life and are associated with significant complications, such as alloimmunisation, allergic reactions, infections, autoimmune reactions and iron overload, which can cause significant morbidity and mortality.4,11,14,15,26-30 Treatment of chronic anaemia places a considerable additional strain on the finite blood supply with blood service organisations facing pressure to maintain sufficient blood supplies and meet demands.31
The impact of transfusions and iron overload in MDS patients

Doctor David Valcárcel, University Hospital Vall d’Hebron
In this video, Dr Valcárcel outlines the risks associated with red blood cell transfusions in patients with MDS. Dr Valcárcel also describes the subsequent effect of iron overload and the impact of iron chelation therapy.
4-minute video
Treating iron overload
Iron overload occurs when iron accumulates in the body and can lead to the production of reactive oxygen species, fibrosis and end organ damage.32,33 Although red blood cell transfusions are the primary cause of iron overload, patients with haematological diseases associated with ineffective erythropoiesis can experience iron overload even in the absence of red blood cell transfusions.12,32,34 Ineffective erythropoiesis can lead to iron overload by reducing hepcidin production, the hormone responsible for regulating iron absorption, which results in increased intestinal iron absorption and increased release of recycled iron from cells into the bloodstream.16,32,35 Iron is usually transported in the bloodstream bound to transferrin, however, in cases of iron overload resulting from regular red blood cell transfusions and/or ineffective erythropoiesis, transferrin becomes saturated leading to an increase in free, non-transferrin-bound iron.36 This excess free iron, which is toxic, accumulates in the body as there is no physiological pathway to excrete it. As the levels of free, non-transferrin-bound iron in the blood increase, iron deposits in various organs leading to clinical complications and organ damage.16,37-39
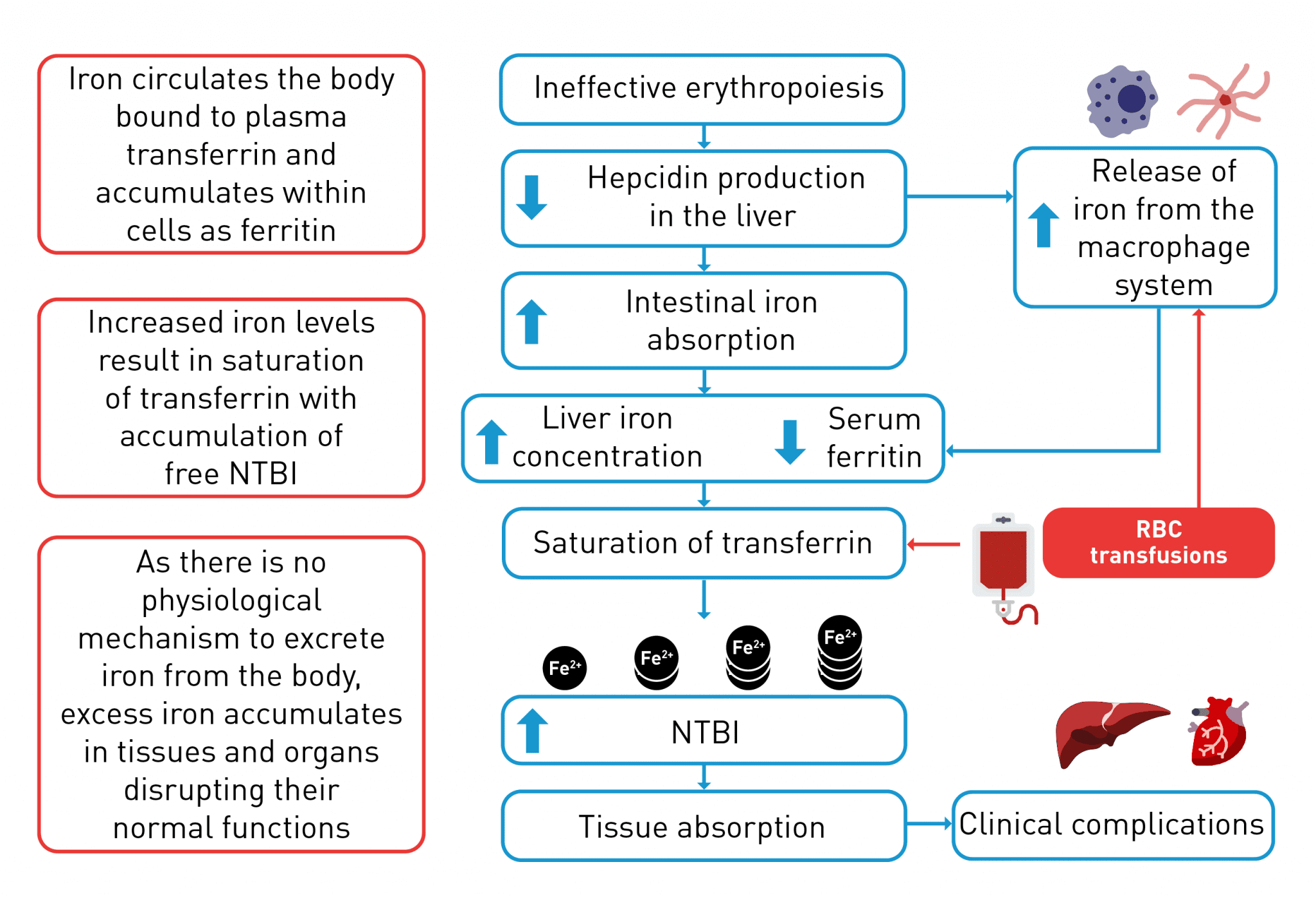
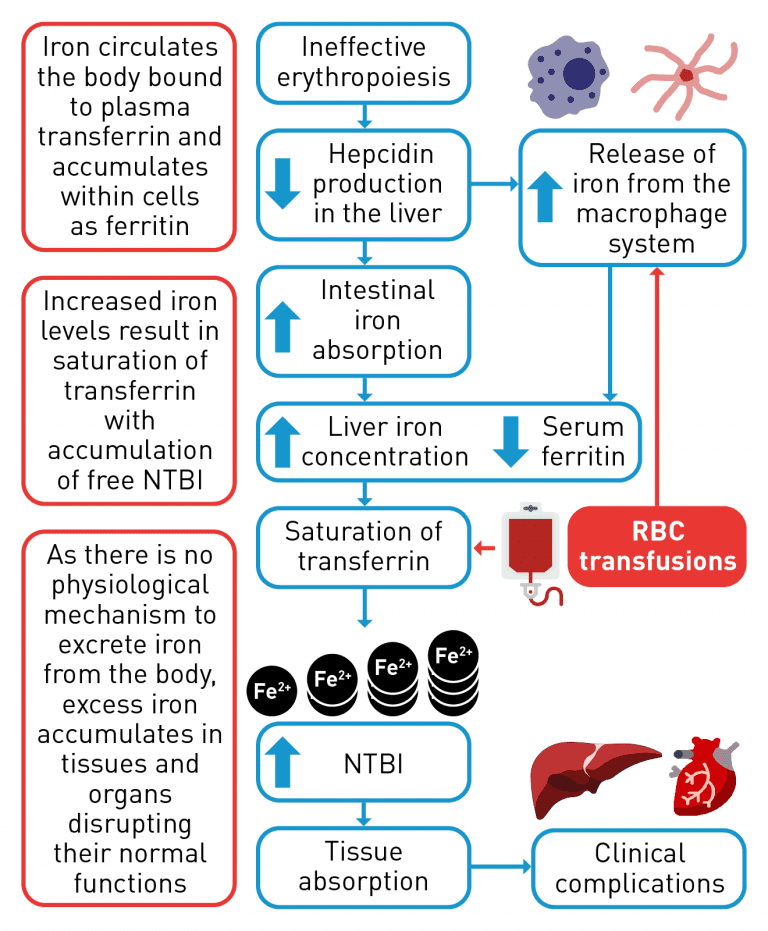
Iron overload occurs when iron accumulates in the body. Additional to the accumulation of iron caused by red blood cell transfusions, ineffective erythropoiesis can lead to iron overload by decreasing hepcidin production in the liver.15,16,32,37-39
NTBI, non-transferrin-bound iron.
Please see Normal and ineffective erythropoiesis for further details
Iron chelation therapy is used to treat iron overload in patients with haematological diseases; iron chelator molecules bind to the free, non-transferrin-bound iron in the bloodstream and allow subsequent excretion via the urine or faeces.15 Iron chelation therapy can balance iron accumulation with iron excretion and prevent increased free, non-transferrin-bound iron concentrations and end organ damage.15 Iron chelation therapy has been shown to be effective in improving survival and decreasing risk of heart failure and morbidities from iron overload. Although iron chelation therapy has markedly improved outcomes in patients, issues still remain, including patient adherence to therapy, cost, potential toxicities and patient burden.40
The unmet need in the treatment of chronic anaemia
For many haematological diseases, the only potential definitive cure is stem cell transplant, which is limited to a small number of eligible patients. Gene therapy may be potentially curative for patients with β-thalassaemia, but as with stem cell transplant, gene therapy is only appropriate for a specific subset of patients.3,7,15,23 Other treatments, such as ESAs and red blood cell transfusions with iron chelation therapy have limitations due to their own complications.1,4,11 ESAs can be effective in patients without adverse prognostic factors, however, many patients with lower-risk MDS receiving ESAs either relapse after an initial response or have primary resistance to these agents; for these patients there are limited effective treatment options available after the failure of ESAs.41 Red blood cell transfusions may result in iron overload and other complications, whilst iron chelation therapy poses certain challenges such as poor adherence and regular monitoring.14,33,37,40 Therefore, in haematological diseases there is a significant unmet need for therapies targeting alternative pathways involved in the development of chronic anaemia, such as those implicated in ineffective erythropoiesis caused by an erythroid maturation defect, with the goal of reducing patient reliance on red blood cell transfusions and iron chelation therapy.
Please see Erythroid maturation defect (EMD) for further details